Quelques définitions et références juridiques
L’article R 5125-9 du Code de la Santé Publique concernant les conditions minimales d’installation pour les officines précise que l’officine doit comporter un emplacement adapté et réservé à l’exécution et au contrôle des préparations magistrales et officinales. Les locaux doivent respecter les recommandations éditées lors de la parution des Bonnes Pratiques de Préparation 2023 publiées par l’ANSM et en vigueur depuis le 20/09/2023.
Ces Bonnes Pratiques de Préparation (BPP) se basent sur une analyse de risque. Elles sont accessibles en ligne (Bonnes Pratiques de Préparation 2023). Des annexes (Analyse pharmaceutique et réglementaire de la prescription, dossier de préparation) sont à consulter pour votre pratique (Bonnes pratiques de préparation – ANSM).
Préparations magistrales, préparations officinales (Article L5121-1)
Préparation magistrale : tout médicament préparé selon une prescription médicale destinée à un malade déterminé lorsqu’il n’existe pas de spécialité pharmaceutique adaptée ou disponible, y compris du fait de l’absence de commercialisation effective, disposant d’une autorisation de mise sur le marché, de l’une des autorisations ou d’un cadre de prescription compassionnelle mentionnés aux articles L. 5121-9-1, L. 5121-12 et L. 5121-12-1, d’une autorisation d’importation parallèle ou d’une autorisation d’importation délivrée à un établissement pharmaceutique dans le cadre d’une rupture de stock d’un médicament, soit extemporanément en pharmacie, soit dans les conditions prévues à l’article L. 5125-1 ou à l’article L. 5126-6.
Préparation officinale : tout médicament préparé en pharmacie, inscrit à la pharmacopée ou au Formulaire National et destiné à être dispensé directement aux patients approvisionnés par cette pharmacie. Le Formulaire National a été revu en 2012, est réactualisé régulièrement et est consultable sur le site de l’ANSM.
Préparations officinales spéciales : à titre exceptionnel et temporaire, pour faire face à la rupture de stock d’un médicament d’intérêt thérapeutique majeur ou à l’arrêt de sa commercialisation ou pour faire face à une menace ou à une crise sanitaire grave et pour garantir la qualité et la sécurité d’utilisation des produits, le ministre chargé de la santé autorise par arrêté la réalisation, par les officines disposant de l’autorisation mentionnée au second alinéa de l’article L. 5125-1-1, pour leur propre compte ou pour le compte d’une autre officine dans les conditions prévues au deuxième alinéa de l’article L. 5125-1,
Préparations pouvant présenter un risque pour la santé
Arrêté du 14 novembre 2014 fixant la liste des préparations pouvant présenter un risque pour la santé :
- Préparations stériles sous toutes formes.
- Préparations à base d’une ou plusieurs substances classées CMR (cancérogènes, mutagènes ou toxiques pour la reproduction). Ex. : de l’acide borique contenu dans le cérat de Galien.
- Préparations destinées aux enfants de moins de 12 ans et contenant une ou plusieurs substances vénéneuses inscrites en liste I, II ou des substances stupéfiants ou psychotropes, à l’exclusion des préparations destinées à être appliquées sur la peau contenant des substances inscrites en liste I ou II.
Leur délivrance ne peut être faite qu’au profit d’une personne physique ou morale connue du pharmacien ou justifiant de son identité.
Le reçu ou la commande :
- doit mentionner le nom des substances, leur quantité, le nom et l’adresse de l’acquéreur,
- l’usage auquel elles sont destinées si la profession de l’acheteur n’implique pas leur emploi,
- est conservé pendant trois ans par le pharmacien pour être présenté à toute réquisition de l’autorité compétente.
La cession des produits toxiques, cancérogènes, tératogènes ou mutagènes (CMR) à une personne âgée de moins de dix-huit ans est interdite (Art. R5132-58)
Exécution ou sous-traitance des préparations pouvant présenter un risque pour la santé
Décret n°2014-1367 du 14 novembre 2014 relatif à l’exécution et à la sous-traitance des préparations magistrales et officinales.
L’exécution de préparations pouvant présenter un risque pour la santé est soumise à une autorisation du directeur général de l’Agence Régionale de Santé (ARS) après demande préalable d’autorisation (article R. 5125-33-1 du CSP).
L’autorisation n’est accordée qu’après enquête d’un inspecteur de l’Agence Régionale de Santé et précise alors les formes pharmaceutiques et les activités autorisées.
L’autorisation de sous-traitance de ces préparations est soumise aux mêmes procédures (cf chapitre 7 BPP : activités externalisées : activités de sous-traitance).
Bonnes Pratiques de Préparation (BPP)
Les préparations pharmaceutiques concernées regroupent les préparations réalisées dans les pharmacies hospitalières et les officines de ville. Elles sont destinées à un ou plusieurs malades lorsqu’il n’existe pas de médicament adapté ou que le médicament est indisponible.
La décision d’application de ces Bonnes Pratiques de Préparation a été publiée par l’ANSM et est applicable depuis le 20/09/2023.
Ce texte de référence opposable est destiné à garantir la qualité des préparations pharmaceutiques. Les préparations magistrales et officinales doivent être réalisées en conformité avec ces BPP.
Le détail de ces BPP sera repris plus loin dans ce même chapitre.
Activité de sous-traitance (cf chapitre 7 BPP)
« Une officine peut confier l’exécution d’une préparation par contrat écrit, à une autre officine qui est soumise, pour l’exercice de cette activité de sous-traitance, à une autorisation préalable délivrée par le directeur général de l’Agence Régionale de la Santé »
Dans le cas où un pharmacien d’officine confie à un confrère la réalisation d’une préparation magistrale qu’il n’est pas en mesure de réaliser (procédé spécialisé, problème d’approvisionnement…), les conditions décrites dans les Bonnes Pratiques de Préparation doivent être respectées.
En effet, la sous-traitance n’exonère pas le pharmacien dit donneur d’ordre de sa propre responsabilité.
Ainsi,
- la sous-traitance d’une préparation n’est envisageable que pour la totalité des opérations de préparation (y compris le conditionnement primaire),
- un contrat écrit doit être signé entre les deux parties et doit notamment préciser le type de préparations réalisées (formes pharmaceutiques + délais de réalisation), les contrôles prévus, les modalités de transport,
- le pharmacien donneur d’ordre (dispensateur) et le sous-traitant doivent conserver une trace écrite des demandes et des livraisons,
- Il est conseillé d’écrire une procédure pour la transmission des préparations au sous-traitant. Cette procédure précisera “qui fait quoi et comment”, notamment la vérification de la bonne adéquation entre formule / forme et patient qui reste de la responsabilité du donneur d’ordre (cf annexe I – Analyse pharmaceutique et réglementaire de la prescription). Elle pourra préciser les modalités de transport…,
- les documents afférents à la préparation sont tenus à la disposition du pharmacien donneur d’ordre,
- Le pharmacien donneur d’ordre, qui dispense la préparation, réalise et trace un contrôle à réception de la préparation afin de s’assurer notamment : du bon étiquetage de la préparation ; de la concordance entre la préparation et la prescription ou la commande de préparations pharmaceutiques ; de l’intégrité physique du conditionnement ; du bon respect des conditions de conservation pendant le transport (par exemple : chaîne du froid, abri de la lumière).
Le donneur d’ordre transcrit sur l’ordonnancier des préparations, outre les mentions légales :
- le nom et l’adresse du pharmacien sous-traitant,
- le numéro d’ordre de transcription par ce dernier sur son ordonnancier,
- sur l’étiquetage de la préparation remise au patient, en plus des mentions légales, doivent apparaître :
- le nom et l’adresse du pharmacien sous-traitant avec son propre numéro d’ordre,
- le nom et l’adresse du pharmacien donneur d’ordre avec son propre numéro d’ordre,
- la date de la préparation et les précautions particulières de conservation et d’utilisation,
- la date de péremption.
Le déconditionnement des spécialités
Une spécialité pharmaceutique relevant de la réglementation des substances vénéneuses ne peut faire l’objet d’un déconditionnement par le pharmacien d’officine en vue de son incorporation dans une préparation magistrale. Cette interdiction n’est pas applicable aux spécialités destinées à être appliquées sur la peau.
Second décret d’application de la loi « Talon » n°88-818 du 22 septembre 1982 :
A titre exceptionnel, une préparation magistrale peut être réalisée à partir d’une spécialité pharmaceutique dans le respect des conditions prévues par les bonnes pratiques de préparation (art. R. 5132-8 CSP).
En effet, les pharmaciens d’officine peuvent être confrontés à la nécessité de déconditionner des spécialités pour réaliser des préparations magistrales (notamment en cas d’ajustement thérapeutique en l’absence de spécialités équivalentes).
Les BPP envisagent cette possibilité si plusieurs conditions sont réunies :
- à titre exceptionnel et en lien avec le prescripteur.
- dans le cas où il n’existe pas de spécialité pharmaceutique adaptée. Cela peut être le cas pour une préparation pédiatrique.
- dans le cadre d’une pathologie pour laquelle cette spécialité n’existe pas.
Ce déconditionnement sera toujours réalisé en prenant en compte l’annexe IV des BPP (Points de vigilance autour de l’’utilisation de spécialités pharmaceutiques déconditionnées (liste non exhaustive)). Il faut rester très vigilant par rapport à la loi Talon et à la faisabilité technique, notamment pour les préparations pédiatriques ou autres (réalisation de poudres diluées afin de garantir la pesée et l’uniformité de teneur, utilisation du Mopral princeps contenant des microgranules gastro-résistantes qui ne doivent pas être broyées).
Il est recommandé lors d’une dilution d’utiliser le même diluant que la spécialité déconditionnée.
Responsabilité
Bonnes Pratiques de Préparation – ANSM (20/09/2023).
La préparation est menée sous la responsabilité d’un pharmacien titulaire ou adjoint, par des personnes compétentes et qualifiées (pharmaciens ou préparateurs en pharmacie).
Le pharmacien a la responsabilité de décision de réalisation des préparations et en apprécie la faisabilité selon une analyse de risque. Tout pharmacien qui reçoit une demande de préparations doit réaliser une analyse pharmaceutique en renseignant l’annexe I (Aide à l’analyse pharmaceutique et règlementaire de la prescription de préparations pharmaceutiques).
L’ensemble des activités reliées au processus de fabrication s’inscrit dans le système de gestion de la qualité mis en place dans la structure par le responsable de la démarche Qualité.
Le pharmacien veillera à préciser clairement, si possible par écrit, les attributions du personnel :
- Il veillera à la formation et à l’actualisation des connaissances du personnel.
- Il élaborera un ensemble de règles d’hygiène, sous forme d’un document écrit qui est porté à la connaissance du personnel. Il veillera à la propreté et à la bonne tenue du préparatoire.
- Il réalisera le suivi des appareils de mesure (balances avec timbre sur le côté attestant de la vérification annuelle par un organisme certifié).
- Il assurera la veille documentaire et l’archivage des documents relatifs aux opérations de fabrication.
La documentation nécessaire à la réalisation de préparations
Bonnes Pratiques de Préparation – ANSM (20/09/2023).
La documentation est un outil de transmission et de conservation de l’information, essentiel à la gestion de la qualité.
Les documents utiles sont :
– Documents nécessaires pour la validation de la préparation (annexe I : aide à l’analyse pharmaceutique et réglementaire de la prescription d’une préparation pharmaceutique).
– Procédures et instructions générales
- réception des matières premières avec inscription dans un registre dédié
- entretien des locaux et du matériel
- personnel habilité et consignes destinées à ce personnel.
- modes opératoires pour les fabrications
- procédures de contrôle des préparations
- gestion des anomalies, des réclamations, des rappels, des déclarations de pharmacovigilance
- gestion des déchets – étiquetage
– Registre des matières premières (voir plus loin)
– Dossier de préparation (selon l’exemple de l’annexe II) qui comprend :
Partie 1 : Validité technico-règlementaire de la préparation pharmaceutique
Partie 2 : La préparation pharmaceutique et son procédé
Partie 3 : Spécifications, contrôles et assurance de la qualité de la préparation pharmaceutique
– Une fiche de fabrication reprendra l’ensemble des parties 2 et 3 et constituera le dossier de lot comprenant 5 parties : Préparation / Conditionnement / Contrôle / Libération pharmaceutique / Gestion des anomalies ou retours /réclamations).
Ces documents contiendront tous les éléments essentiels en termes de qualité et de traçabilité de la préparation. C’est ainsi qu’on devra y trouver :
- matière première : n° de lot et fournisseur, dénomination permettant de retrouver la matière première dans le registre des matières premières…
- les quantités pesées (tickets de pesée) ou mesurées.
- les modes opératoires.
- les contrôles (poids, uniformité de masse, aspect physique…).
- nom de la personne ayant effectué la préparation.
- validation par le pharmacien responsable.
– Ordonnancier des préparations
L’ensemble des dispensations de préparations terminées fait l’objet d’une transcription sur un livre-registre (support papier) ou d’un enregistrement sur support informatique
Ces enregistrements chronologiques comportent :
- Un numéro d’ordre chronologique « numéro d’ordonnancier »
- Le nom et l’adresse du prescripteur
- Le nom et l’adresse du patient
- Le nom de la préparation
- La composition qualitative et quantitative avec indication du numéro de lot de la préparation
- Le nombre d’unités délivrées
- L’identification de la personne ayant réalisé la préparation
- La date de délivrance
Locaux, matériel et personnes habilitées
L’officine doit comporter un local, ou une zone, réservé à l’exécution et au contrôle des préparations magistrales et officinales et de taille adaptée à cette activité. Le cas échéant, ce local peut être utilisé de manière non simultanée pour la préparation des doses à administrer (art. R. 5125-9 CSP).
Cet emplacement doit en outre respecter les exigences requises des Bonnes Pratiques de Préparation, notamment :
- Le préparatoire doit avoir une surface suffisante pour éviter les risques de confusion et de contamination lors des différentes opérations de préparation.
- Il est équipé entre autres d’un bac alimenté en eau chaude et froide, d’éléments de rangement, d’un support parfaitement horizontal pour les balances, d’un meuble réservé à la lecture et à la rédaction des documents.
- Les surfaces et notamment les plans de travail sont lisses et facilement nettoyables. Le local est convenablement aéré, éclairé et maintenu à température appropriée. Il faut noter les dates et heures de nettoyage sur un registre pour en assurer la traçabilité (éventuellement le nom de la personne qui l’a effectué).
Le matériel sera maintenu propre et en bon état de fonctionnement selon des procédures adaptées.
Les appareils de mesure font l’objet de contrôles réguliers dont le résultat sera consigné. C’est ainsi qu’un contrôle annuel des balances électroniques est effectué par un organisme agréé.
Le personnel habilité à réaliser les préparations devra respecter des règles d’hygiène.
Pour les préparations de catégorie 1 (cf. Annexe III), l’utilisation d’une tenue adaptée et propre est obligatoire, incluant le port de la charlotte.
Pour les préparations de catégories 2 et 3, le port de la charlotte, l’utilisation de sur-chaussures et d’une sur-blouse ou d’une tenue propre, dédiée et adaptée à la zone de préparation est obligatoire. L’utilisation de cache-barbe est, le cas échéant, recommandée.
Par ailleurs, un rappel sur la nécessité du lavage des mains avant et après chaque préparation pourrait être affiché dans le local du préparatoire.
Matières premières à usage pharmaceutique (MPUP)
Cela concerne les substances actives (SA), excipients et éléments de mise en forme pharmaceutique.
Les matières premières doivent répondre aux spécifications de la Pharmacopée Européenne dernière édition.
Après leur réception, les matières premières sont mises en quarantaine, pour contrôle de conformité, jusqu’à la décision d’acceptation ou de refus. Chaque récipient est examiné pour en vérifier l’intégrité et l’étiquetage. Les matières premières reçoivent un numéro d’ordre d’identification qui est reporté sur le conditionnement primaire.
Contrôle de conformité
Dans le cas de matières premières contrôlées par un établissement pharmaceutique, l’assurance du contrôle est apportée par un certificat d’analyse ou de conformité du lot. Ce certificat n’est pas toujours envoyé avec la commande mais peut être consultable en ligne sur le site du fournisseur. Il est à archiver dans l’officine.
Si l’établissement fournisseur n’est pas un établissement pharmaceutique, il incombe au pharmacien d’officine de contrôler les matières premières selon les monographies de la pharmacopée.
La matière première est alors affectée d’un numéro d’ordre d’identification propre à l’officine qui est porté sur le registre de matières premières et sur l’étiquette du récipient.
La date d’ouverture de la matière première doit être notifiée afin de connaître sa date réelle de péremption (en général 5 ans après ouverture).
Si une spécialité pharmaceutique est utilisée en tant que matière première dans le cas où il n’existe pas de spécialité pharmaceutique permettant l’ajustement du dosage ou de la forme galénique, aucun contrôle de celle-ci n’est exigé.
En l’absence de date de péremption ou de re-contrôle indiquée sur le conditionnement par le fabricant ou le fournisseur, le pharmacien refuse la matière première.
Registre des matières premières (MPUP)
Celui-ci devra comporter :
- la désignation de la matière première
- la quantité reçue et le nombre de contenants
- le nom du fournisseur
- le numéro de lot apposé par le fournisseur.
- la date de réception
- le numéro d’ordre d’identification donné lors de la réception à l’officine (à reporter sur le conditionnement reçu)
- le numéro de référence du contrôle (soit le n° de référence du certificat d’analyse pour les matières premières venant d’un établissement pharmaceutique, soit le n° de référence donné par l’officine lorsque la matière première a dû être contrôlée au niveau de l’officine)
- le bulletin d’analyses reçu ou édité : oui ou non
- l’acceptation ou le refus avec la signature du pharmacien responsable.
Stockage
La constitution d’une échantillothèque n’est pas obligatoire pour les MPUP provenant d’un établissement pharmaceutique.
On veillera à ne pas mélanger plusieurs lots de matière première dans le même récipient.
On sera attentif à une bonne rotation des stocks et à la péremption des matières premières. Si nécessaire, la destruction sera assurée selon les procédures définies.
Le stockage des substances vénéneuses sera assuré selon la législation en vigueur.
Art. R. 5132-26 CSP (modifié par le Décret n°2007-157 du 5 février 2007)
- Les médicaments relevant de la liste I sont détenus dans des armoires ou des locaux fermés à clef et ne contenant rien d’autre, à l’exception des substances dangereuses classées comme très toxiques ou toxiques, en application de l’article L. 5132-2.
- Les médicaments relevant de la liste II sont détenus séparément de tout autre médicament, produit ou substance, à l’exception des substances classées comme nocives, corrosives ou irritantes, en application de l’article L. 5132-2.
- Toutefois, dans les établissements mentionnés à l’article R. 5124-2 et à l’article R. 5142-1, les médicaments et substances, préparations et produits relevant de la liste I et de la liste II sont détenus en un lieu ou un emplacement dont l’accès est réservé au personnel autorisé.
Les dispositions des trois alinéas précédents ne sont pas applicables aux spécialités pharmaceutiques ayant fait l’objet du conditionnement sous lequel elles sont délivrées aux utilisateurs.
Les médicaments mentionnés au présent article sont disposés de façon à ne pas être directement accessibles au public.
Etiquetage des matières premières acceptées
L’étiquette comportera, outre les mentions apposées par le fournisseur, la date de réception dans l’officine et un numéro d’ordre d’identification donné à l’arrivée dans l’officine.
L’étiquetage des substances vénéneuses sera assuré selon la législation en vigueur.
La Ligne Directrice 2 aborde la préparation de médicaments contenant des substances pouvant présenter un risque pour la santé et l’environnement.

• Une analyse du risque est nécessaire afin de s’assurer que les substances sont manipulées dans des conditions adaptées. Ce risque s’évalue notamment en appréciant :
• le danger ou la toxicité intrinsèque de la substance ;
• l’exposition du personnel (contact direct, ingestion, inhalation, effraction, …) ou de l’environnement à cette substance.
Il est attendu de l’employeur la rédaction d’un Document Unique d’Evaluation des Risques (DUER). Le recensement des substances utilisées et le niveau d’exposition sont pris en compte dans le DUER.
La préparation
Analyse pharmaceutique (voir annexes I et II)
Avant d’entreprendre toute préparation, qu’elle soit officinale ou magistrale, un certain nombre de mesures préalables sont à observer.
Lire le texte de la prescription dans sa totalité et s’assurer de sa bonne compréhension sans hésiter sur un mot ou une posologie ;
Vérifier systématiquement :
- si les posologies par prise et/ou par 24h sont respectées,
- si les composants sont inscrits sur les listes des substances vénéneuses,
- si les composants présentent une incompatibilité physico-chimique entre eux ;
Vérifier les quantités demandées et les abréviations telles :
- FSA (Fac Secundum Artem) (Faire selon l’art)
- ââ (ana partes aequales) (à parties égales)
- QS (Quantité Suffisante)
- QSP (Quantité Suffisante Pour)
Ne pas confondre :
- Doses usuelles et doses maximales (Tableaux de posologie, Pharmacopée Française, Xème édition, 1988 et mises à jour du J.O.)
- Doses d’exonération dont il faut tenir compte pour l’étiquetage et la dispensation (“Substances vénéneuses – Listes et exonérations” Les éditions des Journaux Officiels. Mise à jour de mai 2015).
Modifier une dose dépassée après concertation avec le médecin
Attention ! La confirmation par le médecin “je dis…” n’exonère pas le pharmacien de sa responsabilité.
- Vérifier sa conformité à la législation (notamment aux décrets pris en application de la loi dite TALON)
- Résoudre les éventuels problèmes d’ordre pharmaceutique et galénique
- Réflexion sur les excipients à effet notoire (EEN) : il appartient au pharmacien de vérifier auprès du patient qu’il ne présente pas de contre-indications vis-à-vis de ces excipients (déficit enzymatique, diabète, allergie…). La liste des EEN est mise à jour régulièrement et est consultable sur le site d’ANSM.
- En ce qui concerne les pommades, il faut être très vigilant sur la faisabilité de la préparation (mélange de phases non miscibles, oxydation des SA..). Il existe sur le marché des « excipients pour préparations magistrales ». Ceux-ci sont parfois assez spécifiques pour certaines SA et il est important de consulter les sites des laboratoires afin de vérifier les compatibilités excipient-SA.
- Utiliser un procédé technique permettant de résoudre l’incompatibilité. Ceci est du ressort du pharmacien qui doit pouvoir, sans en référer au médecin, utiliser des procédés galéniques tels que :
Adjuvants de solubilité : la caféine peut être solubilisée par addition de benzoate de sodium et l’iode par addition d’iodure de potassium…
Poudres inertes : elles sont utilisées pour éviter la formation de mélanges pâteux entre deux substances pulvérulentes, telles l’aspirine et le bicarbonate de sodium… Un exemple est la silice colloïdale.
Sels au lieu de bases : les sels d’alcaloïde sont généralement solubles en milieu aqueux, la base l’est normalement en milieu alcoolique ou huileux. Il faut alors tenir compte des équivalences. (Ex. : phosphate de codéine à la place de codéine base).
Addition de lanoline (EEN) ou de tout autre excipient amphiphile :dans une préparation dermatologique à base de vaseline contenant une phase aqueuse, sans pour autant en modifier la concentration en substance active.
L’évaluation des risques
Le tableau ci-dessous permet de classer les préparations selon une analyse de risque en fonction de la ou les SA, de la voie d’administration, des opérations pharmaceutiques et du nombre de patients susceptibles de recevoir un lot de préparations (cf Annexe III).
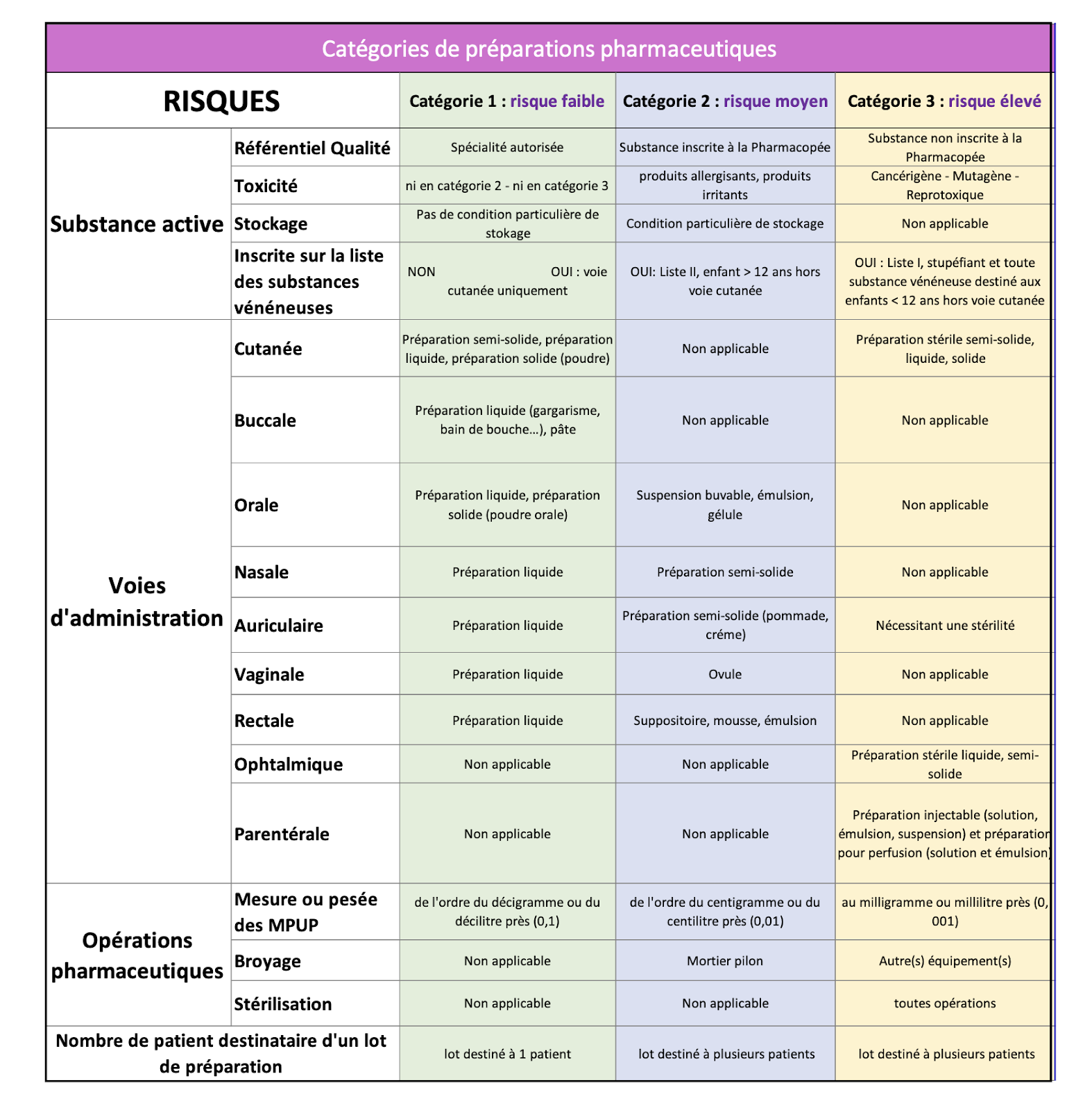
La préparation (Annexe II : exemple de dossier de préparation)
Quelques règles essentielles :
- Une seule préparation à la fois.
- Une seule personne pour réaliser une préparation.
- Ne pas interrompre avant la réalisation complète de la préparation.
- Les consignes par écrit sont à effectuer au moment où chaque action est menée.
Le manipulateur respecte les règles d’hygiène (lavage des mains, gants, coiffe et masque si nécessaire) et s’assure de la propreté du local (paillasse…) et du matériel.
Il suit les règles d’ordre de mélange des produits lors de la préparation (du plus toxique au moins toxique, de la plus petite quantité à la plus importante, règle de miscibilité…).
Il vérifie que le matériel utilisé pour les pesées est de portée et de sensibilité adaptées aux masses mesurées. Il tient compte de la précision de la balance.
- Soit d l’échelon réel correspondant à la précision de l’affichage et à la résolution de la balance (d= 0,1, d= 0,01, d= 0,001, d= 0,0001)
- Soit m la masse à peser
Pourcentage d’erreur sur la pesée = d/m X100
Pour la pesée d’une matière première, on considère que ce pourcentage ne doit pas excéder 1%. Il faudra néanmoins prendre en considération la toxicité du produit à peser. S’il s’agit d’une pesée destinée à un contrôle analytique, ce pourcentage ne doit pas excéder 0,1%.
Toutes les mesures de pesée ou de volume sont enregistrées et reportées dans la fiche de fabrication, l’édition d’un ticket de pesée étant recommandée.
Les modalités de préparation sont également consignées au fur et à mesure de leur déroulement. Les récipients sont ouverts et refermés en évitant toute contamination croisée avec d’autres matières premières et toute contamination microbienne.
Conditionnement et étiquetage
Le conditionnement sera adapté à la forme galénique réalisée, aux conditions de stabilité des composants et son état de propreté sera vérifié.
Sans préjudice des dispositions de l’article R. 5132-15, l’étiquetage du conditionnement primaire et, s’il existe, du conditionnement extérieur des préparations mentionnées aux 1° à 3° de l’article L. 5121-1 porte, sur fond blanc, les mentions suivantes, inscrites de manière à être facilement lisibles, clairement compréhensibles et indélébiles :
Mentions relatives à l’identification de la préparation
a) Le nom ou la dénomination de la préparation, le dosage, la forme pharmaceutique et, le cas échéant, la mention du destinataire ( » nourrissons « , » enfants « , » adultes » ou, le cas échéant, toute autre catégorie de patients dont les caractéristiques nécessitent une mention particulière) ;
b) La composition qualitative et quantitative en substances actives par unité de prise ou, selon la forme d’administration, pour un volume ou une masse déterminé, en utilisant, le cas échéant, les dénominations communes ;
c) Le contenu en masse, en volume ou en unités de prise ;
d) Lorsqu’il s’agit d’une préparation sous forme liquide, les mentions du b et du c sont remplacées par la quantité totale de chaque substance active dans le volume total de solution et la concentration en unité de masse par volume ;
e) Les excipients qui ont une action ou un effet notoire. Toutefois, s’il s’agit d’un produit injectable, d’une préparation topique ou d’un collyre, tous les excipients sont mentionnés ;
f) La voie d’administration si le produit est destiné à être administré directement au patient.
Pour les préparations qui ne sont pas destinées à être administrées directement au patient et qui sont utilisées pour la réalisation d’autres préparations, l’étiquette comporte dans un encadré rouge et en caractères rouges la mention : “Ne pas administrer – Réservé à la réalisation de préparations en pharmacie” ainsi que les modalités d’utilisation ;
g) Le mode d’administration, si nécessaire ;
h) Lorsque la préparation est destinée à une autre voie d’administration que la voie orale, sublinguale ou perlinguale, l’étiquette porte la mention “Ne pas avaler” en caractère gras et noirs sur fond rouge.
Mentions relatives au numéro de lot et à la traçabilité :
a) Le numéro du lot de la préparation réalisée par une officine, une pharmacie à usage intérieur ou un établissement pharmaceutique. Lorsque celle-ci est réalisée dans les conditions de sous-traitance mentionnées aux articles L. 5125-1 et L. 5126-2, le numéro du lot est celui de l’officine, de la pharmacie à usage intérieur ou de l’établissement pharmaceutique ayant réalisé la préparation ;
b) Le numéro d’enregistrement de la préparation figurant sur le livre-registre ou dans le système informatisé ;
c) La date limite d’utilisation ;
d) Les précautions particulières de conservation, s’il y a lieu ;
e) Le nom et l’adresse de la pharmacie à usage intérieur ou de la pharmacie d’officine ayant réalisé et dispensé la préparation ou le nom et l’adresse de l’établissement pharmaceutique ayant réalisé la préparation. Lorsque ladite préparation est réalisée pour le compte d’une pharmacie à usage intérieur ou d’une officine, le nom et l’adresse de la pharmacie à usage intérieur ou de l’officine ayant dispensé la préparation sont également mentionnés sur l’étiquette ou sur une contre-étiquette.
Remarques :
Pour établir une date de péremption (date limite d’utilisation) pour une préparation non stérile, les règles suivantes peuvent être appliquées à titre indicatif.
- pour les formules liquides non aqueuses et les solides :
– quand une spécialité pharmaceutique est utilisée comme source de substance active de la préparation pharmaceutique : la date de péremption ne dépasse pas la date la plus rapprochée des deux suivantes :
o soit 25% du temps restant jusqu’à la péremption de la spécialité utilisée ;
o ou ne dépassant pas 6 mois.
- pour les formules contenant de l’eau :
– formules préparées avec des ingrédients de forme solide ou liquide :
ce type de préparation pharmaceutique effectuée sans information ou donnée sur sa stabilité constitue un risque élevé pour le patient ;
la date de péremption ne dépasse pas 14 jours pour les préparations liquides conservées entre 2°C et 8°C. Elle sera inférieure à 14 jours si elle est conservée à température ambiante.
La préparation de gélules ou de sachets peut être privilégiée.
- pour les autres formules :
– la date de péremption est la date la plus proche de :
la date de fin de traitement ;
ou 30 jours maximum.
En cas de sous-traitance, cette date de péremption peut être augmentée de 72h afin
de prendre en compte les délais d’acheminement des préparations et la continuité du traitement.
Etiquetage des préparations

Contrôle de la préparation (Annexe II – Partie 3)
Généralités
Toute préparation magistrale ou officinale terminée, encore appelée “produit fini”, doit satisfaire à certains contrôles avant sa dispensation au patient. Les essais à effectuer et les normes à respecter sont précisés dans la Pharmacopée Européenne 11ème édition :
- Monographie des formes pharmaceutiques
- Essais de pharmacotechnie
Le pharmacien d’officine doit veiller à réaliser des contrôles sur les formes fabriquées puisqu’il engage sa responsabilité. Seul un pharmacien au sein de l’officine est apte à procéder à la libération des préparations suite aux résultats de ces contrôles. La signature du pharmacien doit être apposée sur la fiche de fabrication et l’ordonnancier.
Les contrôles effectués à l’officine sont simples mais restent efficaces et non destructeurs, compte tenu des petites quantités fabriquées.
Le pharmacien doit toujours garder à l’esprit que ces contrôles garantissent :
- la qualité du produit (dosage, homogénéité, stabilité)
- la quantité du produit correspondant au traitement total prescrit.
- la traçabilité (ordonnancier, étiquetage, fiche de fabrication)
A titre d’exemples, quelques essais pharmacotechniques sont proposés ci-dessous ; la liste, sans être exhaustive, est représentative de ce qu’il est possible de faire en officine sans problème particulier.
Préparations solides pour la voie orale
Les gélules
Les gélules doivent être parfaitement fermées et le pharmacien doit vérifier :
- le nombre d’unités délivrées.
- l’homogénéité de la poudre contenue si mélange de poudres. Afin d’assurer cette homogénéité lors de la réalisation du mélange, on peut utiliser un traceur coloré inscrit à la Ph. Eur.
- Pour les formes pharmaceutiques unitaires, une uniformité de masse est réalisée selon les normes de la Pharmacopée ou selon une méthode adaptée et équivalente à la Pharmacopée. Pour les préparations destinées à être stockées, ou lorsqu’un lot de préparations est destiné à plus de 10 patients, une uniformité de teneur est réalisée.
L’essai d’uniformité de masse des préparations présentées en unités de prises de la Pharmacopée Européenne (chap. 2.9.5.) est destructif car il nécessite d’ouvrir les gélules. Il est possible alors de réaliser un essai non destructif d’uniformité de masse des gélules préparées.
Exemple donné pour 20 unités :
- pesée des 20 gélules vides servant à la préparation = M1
- M1/20 = masse moyenne d’une enveloppe = m1
- remplissage des 20 gélules
- pesée des 20 gélules remplies de poudre = M2
- M2 – M1 = M3 = masse totale de la poudre
- M3/20 = m = masse moyenne de poudre par gélule
Peser individuellement et précisément les 20 gélules pleines ;
Retirer de chaque pesée individuelle m1, on obtient la masse de poudre contenue dans chaque gélule.
La masse de poudre contenue dans chaque gélule est comparée à la masse moyenne de poudre. Cet essai reflète la qualité du remplissage mais ne permet pas de conclure à une conformité à la Pharmacopée puisque l’essai est différent. La conclusion porte sur l’acceptation ou le refus des gélules préparées.
Les valeurs à retenir sont les suivantes :
| masse moyenne = m | écart limite en % de la masse moyenne |
| moins de 300 mg | ± 10 |
| 300 mg et plus | ± 7,5 |
Les paquets
Il faut vérifier :
- Le nombre d’unités et leur propreté,
- L’homogénéité de pliage et de taille,
- L’homogénéité de la poudre contenue,
- La masse unitaire.
Pour une poudre en unités de prise (cas des paquets)
| masse moyenne = m | écart limite en % de la masse moyenne |
| moins de 300 mg | ± 10 |
| 300 mg et plus | ± 7,5 |
Préparations liquides pour la voie orale
- Masse ou volume délivrable,
- Uniformité de masse de la dose délivrée par les récipients multidoses
- Homogénéité
- Limpidité pour les solutions
Dans le cas des suspensions : évaluation de la suspension obtenue :
- Bon écoulement de la suspension hors du flacon
- Homogénéité de la dispersion de la substance active (absence de sédiment ou de précipité ce qui signifie que l’agent de suspension a été bien choisi)
Préparations semi-solides pour application cutanée
- Aspect du tube ou du pot : nombre de plis, propreté…
- Masse unitaire : % d’erreur accepté = ± 10 %
- Homogénéité : dispersion sans agglomérat ou dissolution des P.A. dans la phase adéquate.
Pour toutes les préparations, le contrôle de l’intégrité et de la propreté des conditionnements primaires et secondaires est indispensable, auquel il faut ajouter le contrôle de l’étiquetage.
Le remboursement des préparations
L’étudiant est invité à consulter les mises au point de l’Assurance Maladie sur le site Ameli.
Le décret n°2006-1498 du 29 novembre 2006 et l’arrêté du 20 avril 2007 précisent les dispositions réglementaires encadrant la prise en charge des préparations magistrales et officinales. Il en résulte que la plupart des préparations magistrales et officinales ne sont plus remboursables par l’Assurance Maladie.
La réglementation de la Sécurité Sociale est à appliquer selon la circulaire 58/2008 datant du 05/11/2008.
Les quatre critères pour une prise en charge sont :
- Il y a un objectif thérapeutique réel
- L’intérêt de santé publique est clairement démontré : efficacité établie et reconnue, place majeure dans la stratégie thérapeutique, affection présentant un caractère habituel de gravité
- Il n’existe pas d’alternative utilisant des spécialités ou produits disponibles, remboursables ou non
- Toutes les matières premières rentrant dans la composition de la préparation sont inscrites à la Pharmacopée Européenne
Seules les matières premières simples utilisées à titre d’excipient sont remboursables. Les excipients composés commercialisés ne le sont pas. Leur utilisation ne remet pas en cause le caractère éventuellement remboursable de la préparation, mais leur coût ne doit pas être facturé à l’assurance maladie.
En pratique, les préparations qui restent remboursables se limitent à quelques situations au cas par cas :
- Les préparations destinées à une utilisation en pédiatrie ou en gériatrie pour l’adaptation de posologies dans les cas où il n’existe pas de dosage adapté de la spécialité (y compris à l’hôpital).
- Les préparations à visée dermatologique pour des patients atteints de maladies rares, orphelines, génétiques à expression cutanée ou chroniques particulièrement graves et étendues. A titre exceptionnel, tous les excipients – simples et composés – utilisés dans les préparations dermatologiques spécifiques à certaines pathologies sont pris en charge.
Certains cas particuliers :
- Pathologies rénales (acidoses métaboliques chroniques) ou dialyses : bicarbonate de sodium, chlorure de sodium et carbonate de sodium en comprimés ou gélules ;
- Mucoviscidose : bêta-carotène en comprimés ou gélules ;
- Oncologie : préparations d’associations remboursables en bain de bouche.
Si le médecin considère que la préparation respecte les règles de prise en charge par l’Assurance Maladie, il doit alors écrire sur l’ordonnance « Prescription à but thérapeutique en l’absence de spécialités équivalentes disponibles » engageant ainsi sa responsabilité. En l’absence de cette mention, la préparation ne peut pas être facturée.
Si le médecin a porté cette mention mais qu’à l’issue de l’analyse pharmaceutique, il apparaît que la préparation ne fait partie des préparations remboursables, elle ne doit pas davantage être présentée au remboursement.
Ce n’est donc que si le médecin a bien inscrit cette mention et que la préparation respecte les critères de prise en charge prévus que le pharmacien pourra la facturer à l’Assurance Maladie, en utilisant le code adapté à son taux de prise en charge (essentiellement PMR ou PM4).
Bibliographie
- Code de la Santé Publique et Code de déontologie – Consultable également sur le site Légifrance.
- Code de la Sécurité Sociale.
- ANSM : site et publications.
- Ordre des Pharmaciens : site et publications.
- Assurance Maladie : site Ameli.
- Pharmacopée européenne – 11ème édition.
- Substances Vénéneuses – Listes et exonérations- 2015- Les éditions du Journal Officiel, Paris.
- Bonnes Pratiques de Préparation. Publication à l’ANSM (20/09/2023).
- A. Le Hir, J.-C. Chaumeil, D. Brossard, C. Charrueau, S. Crauste-Manciet. Pharmacie Galénique. Bonnes pratiques de fabrication- 10ème édition- 2016- Editions Masson, Issy-les-Moulineaux.
- P. Wehrlé et coll. Pharmacie galénique- Formulation et technologie pharmaceutique- 2007- Editions Maloine, Paris.
- C. Mautrait et R. Raoult. La préparation : mode d’emploi- 2ème édition- 2009- Editions du Porphyre, Paris.
- J.-M. Fonteneau et P. Klusiewicz. TP de préparation et de conditionnement des médicaments- 2ème édition- 2014- Editions du Porphyre, Paris.