Introduction
Si le pharmacien peut préparer et dispenser des médicaments, il peut aussi faire commerce de marchandises figurant sur une liste arrêtée par le ministre chargé de la santé (art. L. 5125-24 CSP), tels que les produits cosmétiques, les dispositifs médicaux ou encore les compléments alimentaires. Chacun de ces produits bénéficiant d’un cadre juridique particulier dans le but de la protection de la santé publique, tout pharmacien doit pouvoir correctement qualifier les produits qu’il conseille et dispense aux patients.
Il doit savoir déterminer, en toutes circonstances, si un produit donné est ou n’est pas un médicament. La maîtrise de cette distinction, souvent moins évidente qu’il n’y paraît au premier abord, est primordiale. En effet, un manque de rigueur ou simplement d’attention peut conduire à des erreurs de présentation ou de dispensation, sources de risques pour le consommateur et sera susceptible d’engager la responsabilité civile, pénale ou disciplinaire des professionnels du médicament.
La définition du médicament
Cette définition figure à l’article L. 5111-1 du Code de la santé publique. Elle est issue d’une loi de transposition du Code communautaire du médicament à usage humain (directive européenne 2001/83/CE du 6 novembre 2001 modifiée). Ainsi, le médicament est défini de la façon suivante : “On entend par médicament toute substance ou composition présentée comme possédant des propriétés curatives ou préventives à l’égard des maladies humaines ou animales ainsi que toute substance ou composition pouvant être utilisée chez l’homme ou chez l’animal ou pouvant leur être administrée, en vue d’établir un diagnostic médical ou de restaurer, corriger ou modifier leurs fonctions physiologiques en exerçant une action pharmacologique, immunologique ou métabolique. Sont notamment considérés comme des médicaments les produits diététiques qui renferment dans leur composition des substances chimiques ou biologiques ne constituant pas elles-mêmes des aliments, mais dont la présence confère à ces produits, soit des propriétés spéciales recherchées en thérapeutique diététique, soit des propriétés de repas d’épreuve. »
L’interprétation extensive de la notion de médicament a notamment pour but de protéger les consommateurs de produits de santé. En outre « lorsque, eu égard à l’ensemble de ses caractéristiques, un produit est susceptible de répondre à la fois à la définition du médicament (…) et à celle d’autres catégories de produits » tels que les cosmétiques ou les compléments alimentaires, il sera considéré comme un médicament.
Les différentes catégories de médicaments à l’officine
L’officine peut détenir et dispenser deux catégories de médicaments : des spécialités pharmaceutiques et des préparations magistrales ou officinales.
NB : Jusqu’en juillet 2016, les officines pouvaient dispenser des médicaments dits « Produits officinaux divisés » (POD). Ces médicaments étaient des « drogues simples, produits chimiques ou préparations stables décrites par la pharmacopée, préparés à l’avance par un établissement pharmaceutique et divisés soit par lui, soit par la pharmacie d’officine qui le met en vente ». Cette catégorie de médicament a été supprimée du CSP car non conforme au droit européen.
En pratique, les POD se présentaient souvent sous forme de sachets préconditionnés (ex: sulfate de magnésium, acide borique etc…) qui pouvaient être dispensés en l’état pour un usage thérapeutique.
Les spécialités pharmaceutiques
L’article L.5111-2 du CSP les définit comme « tout médicament préparé à l’avance, présenté sous un conditionnement particulier et caractérisé par une dénomination spéciale ». Plus précisément, toute spécialité pharmaceutique – ou médicament fabriqué industriellement ou selon un processus industriel – doit avoir bénéficié d’une autorisation de mise sur le marché, obtenue auprès de l’Agence européenne des médicaments (EMA) ou de l’Agence nationale de sécurité du médicament et des produits de santé (ANSM) (Article L.5121-8 CSP).
En outre, l’article 47 du Code de Déontologie (article R.4235-47 CSP) interdit au pharmacien de délivrer un médicament non autorisé. Le conditionnement, l’étiquetage et la notice des spécialités pharmaceutiques doivent concourir à l’information des patients et au bon usage des médicaments (articles R.5121-138 à 140, R.5121-202, R.5132-15 CSP).
Les préparations magistrales et officinales (L.5121-1 du CSP)
Ce sont des médicaments fabriqués dans l’officine ou sous traités à une officine habilitée.
(cf. Chapitre Les préparations à l’officine)
Les spécialités pharmaceutiques
Codification des spécialités pharmaceutiques
Le Code Identifiant de Présentation
Historiquement, le « code CIP » (Code Identifiant de Présentation) a été créé par le Club Inter-Pharmaceutique (regroupant des représentants de l’officine, des pharmaciens hospitaliers, des distributeurs en gros et des industriels) pour identifier les médicaments. Depuis le 1er janvier 2009 le code identifiant de présentation (code CIP) à 7 chiffres est remplacé par un code à 13 chiffres. Ce code à 13 chiffres est mentionné dans la décision d’autorisation de mise sur le marché de toute spécialité pharmaceutique.
NB : Les produits sans AMM ont été regroupés dans la codification ACL (Association de Codification Logistique).
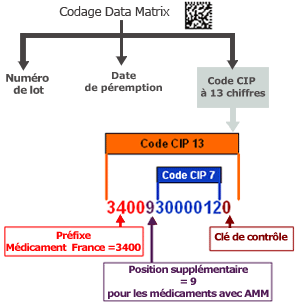
Le code DataMatrix
De plus, suite aux évolutions réglementaires concernant la traçabilité des médicaments, le conditionnement des spécialités pharmaceutiques comporte un marquage GS1 DataMatrix™, intégrant le code CIP 13, le numéro de lot et la date de péremption (obligatoire depuis 2011). Cette traçabilité permet de renforcer la sécurité de la chaîne du médicament, de lutter contre la falsification et d’améliorer la gestion des stocks, depuis les établissements pharmaceutiques de fabrication jusqu’aux officines.

Ce mécanisme peut aussi faciliter les rappels de lots de médicaments, présents dans les officines ou au domicile des patients, grâce au dossier pharmaceutique. Enfin, les informations du numéro de lot et de la date de péremption sont portées sur un document de traçabilité accompagnant la livraison par les distributeurs en gros aux officines (à conserver 5 ans).
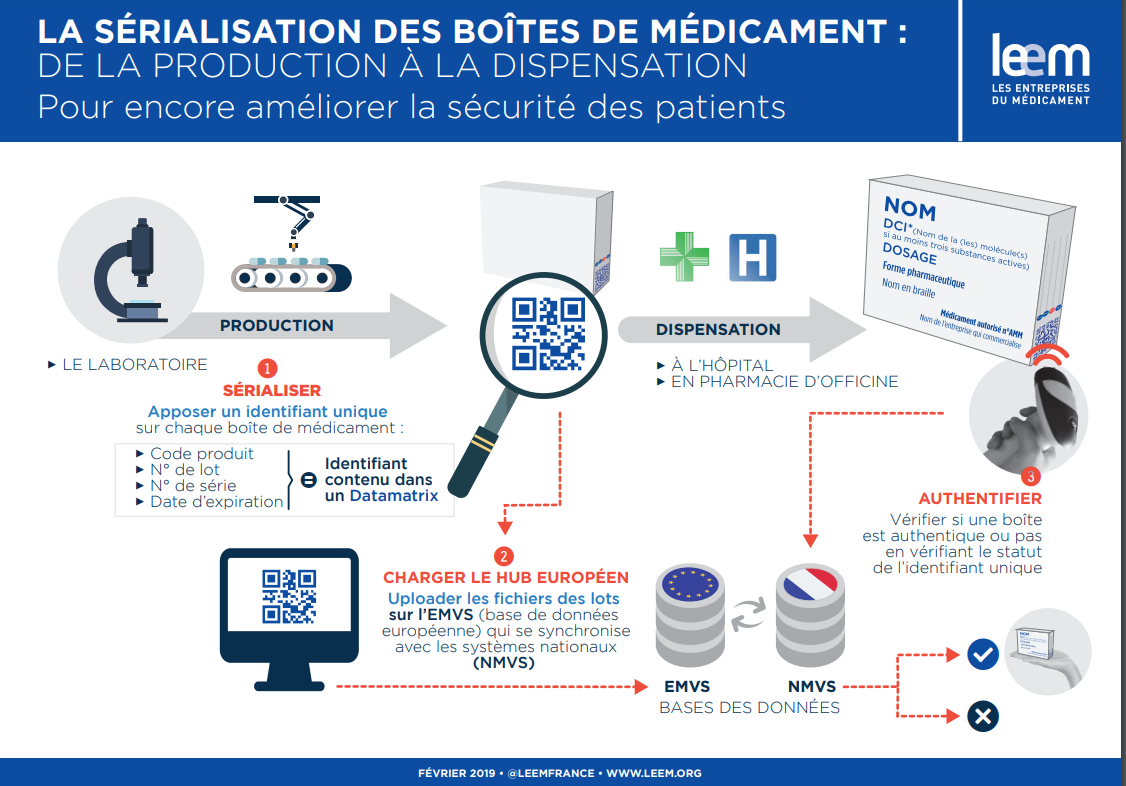
La sérialisation de spécialités pharmaceutiques
En juin 2011, l’Union européenne a adopté la directive 2011/62/UE relative à la prévention de l’introduction dans la chaîne d’approvisionnement légale de médicaments falsifiés. Ce texte introduit notamment des mesures de sécurité pour la dispensation de médicaments à l’échelle européenne et prévoit de doter certains médicaments de « dispositifs de sécurité » (safety features).
Ces dispositifs de sécurité sont de deux types :
- Un dispositif anti-effraction pour tous les médicaments humains ;
- Un identifiant unique (sérialisation), correspondant à un numéro de série pour tous les médicaments de prescription obligatoire, ainsi que quelques rares médicaments de prescription facultative présentant un risque particulier de falsification (ceux figurant à l’annexe II du règlement délégué).
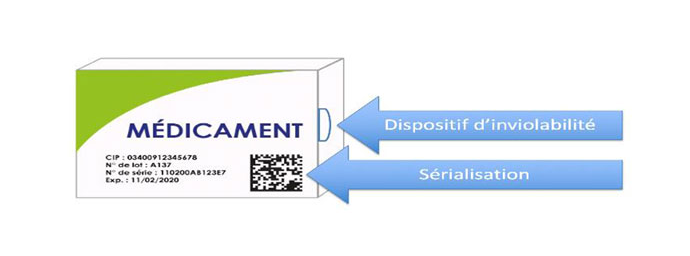
Le contrôle des médicaments sérialisés est obligatoire en officine avec un objectif fixé par l’Etat de 100% des pharmacies raccordées au 31 décembre 2022.
France MVO (France Medicines Verification Organisation) est l’organisme national de gouvernance de l’authentification des médicaments à usage humain.
Consulter le cahier thématique de l’ONP : L’authentification des médicaments à usage humain
Etiquetage des spécialités pharmaceutiques
L’étiquetage du conditionnement extérieur ou à défaut du conditionnement primaire d’une spécialité pharmaceutique doit porter certaines mentions qui peuvent être utiles lors de la dispensation (art. R.5121-138 CSP, art.R.5121-140 CSP).
L’emballage extérieur comporte un espace blanc, entouré d’un filet coloré, dans lequel le pharmacien ou le médecin dispensateur inscrit la posologie prescrite. Le filet coloré est rouge pour les médicaments relevant de la liste I, vert pour ceux qui relèvent de la liste II.
Le conditionnement extérieur peut comporter, en outre, des signes ou des pictogrammes complétant certaines informations (conservation, péremption, conduite à tenir…).

Le Triangle noir inversé (« black symbol »)
Il symbolise la mise sous “surveillance renforcée” du médicament lorsque :
- Peu d’informations sont disponibles à son sujet, par exemple au début de sa commercialisation,
- Il existe des informations limitées concernant son utilisation prolongée.
Il est utilisé dans tous les Etats Membres de l’Union européenne et figure dans la notice d’information des patients et dans le résumé des caractéristiques du produit (RCP).
Cela ne signifie pas que le médicament pose des problèmes de sécurité.

«Ce médicament fait l’objet d’une surveillance supplémentaire.»
Depuis le 25 avril 2013, une liste européenne de médicaments sous surveillance renforcée est publiée tous les mois par l’Agence européenne du médicament (EMA).
Consultez: Médicaments sous surveillance renforcée (AMSM)
Les « marques ombrelles »
On désigne sous le nom de « marque ombrelle », un même nom de marque qui porte sur des produits très hétérogènes. Largement utilisée pour les produits de consommation courante (un même nom de marque peut être utilisé pour des pâtes alimentaires et des sauces par exemple), cette stratégie marketing est susceptible de tromper le patient dans le domaine de la pharmacie. En effet, la dénomination commerciale parfois très proche de certains produits de statuts juridiques très différents (marques ombrelles) peut comporter un risque de confusion chez les patients.
En tant que professionnels du médicament, il est important que vous sachiez les distinguer.
Exemples :
- Alodont® (médicament), Alodont® protect (produit cosmétique) et Alodont® fix (dispositif médical)
- Euphytose® (médicament), Euphytose® Zen/Nuit/Confort intestinal (compléments alimentaires)
Afin de limiter ces risques, l’ANSM a émis des recommandations pour les industriels : L’ANSM publie ses recommandations sur les noms des médicaments
Les spécialités génériques
Définition
Art. L. 5121-1 5° CSP : « Sans préjudice des articles L. 611-2 et suivants du Code de la propriété intellectuelle, une spécialité générique d’une spécialité de référence est définie comme celle qui a la même composition qualitative et quantitative en principes actifs, la même forme pharmaceutique et dont la bioéquivalence avec la spécialité de référence est démontrée par des études de biodisponibilité appropriées.
Une spécialité ne peut être qualifiée de spécialité de référence que si son autorisation de mise sur le marché a été délivrée au vu d’un dossier comportant, dans des conditions fixées par voie réglementaire, l’ensemble des données nécessaires et suffisantes à elles seules pour son évaluation.
Pour l’application du présent alinéa, les différentes formes pharmaceutiques orales à libération immédiate sont considérées comme une même forme pharmaceutique. De même, les différents sels, esters, éthers, isomères, mélanges d’isomères, complexes ou dérivés d’un principe actif sont regardés comme ayant la même composition qualitative en principe actif, sauf s’ils présentent des propriétés sensiblement différentes au regard de la sécurité ou de l’efficacité. Dans ce cas, des informations supplémentaires fournissant la preuve de la sécurité et de l’efficacité des différents sels, esters ou dérivés d’une substance active autorisée doivent être données par le demandeur de l’autorisation de mise sur le marché… ».
Autorisation de mise sur le marché (AMM)
Un médicament générique peut être introduit sur le marché dès que la protection du brevet du produit princeps est échue. Cette procédure d’AMM est beaucoup moins longue pour les génériques. La différence réside dans le dossier d’enregistrement, qui peut être allégé, les parties correspondant à la pharmaco-toxicologie et à la clinique pouvant être bibliographiques sauf en ce qui concerne la démonstration de la bioéquivalence.
Le demandeur doit fournir un dossier pharmaceutique et des études de bioéquivalence quand il y a modification de la composition qualitative des excipients.
Les autorités d’enregistrement accordent donc à la spécialité générique les mêmes conditions de P/D, mais aussi un même taux de remboursement que le médicament initial (sous réserve que la spécialité de référence soit inscrite sur la liste des spécialités remboursables) et surtout un prix inférieur (d’au moins 60 % par rapport au PFHT) à celui de la spécialité de référence.
Identification de la spécialité générique
L’ordonnance du 24 avril 1996 subordonne le remboursement des spécialités génériques à une claire identification de ces dernières : peuvent être inscrites sur les listes des médicaments remboursables, les spécialités dont le nom commercial sera constitué :
- Soit par la DCI, assortie d’un nom de marque ou du nom du titulaire de l’AMM ou de l’exploitant ;
- Soit par un nom de fantaisie suivi du suffixe “Gé”, souligné d’un trait.
Répertoire des génériques
Afin de faciliter l’identification des génériques, un répertoire officiel a été créé, le répertoire des génériques, disponible sur le site de l’ANSM. Ce répertoire présente les spécialités génériques et leurs spécialités de référence « princeps » par groupe générique. Les groupes génériques sont regroupés par principe actif (en dénomination commune) et par voie d’administration.
L’article R.5121-5 CSP confie aux soins du directeur général de l’ANSM, l’identification des spécialités répondant à cette définition et leur inscription à ce répertoire. Cette décision ainsi que le répertoire sont publiés sur le site de l’ANSM. Les excipients à effets notoires sont identifiés dans le répertoire par spécialité.

Prescription en DCI – Droit de substitution
Depuis 2009, la prescription libellée en dénomination commune est obligatoire pour les spécialités figurant dans un groupe générique. Face à une telle prescription, le pharmacien dispense une spécialité appartenant à ce groupe.
Si, malgré tout, la prescription est établie en nom commercial, et en l’absence d’opposition du prescripteur, la substitution peut se faire entre génériques d’un même groupe ou entre la spécialité de référence du groupe et un générique de ce groupe (Guide de la CNAMTS).
La délivrance de cette spécialité ne doit pas entraîner une dépense supplémentaire supérieure à la dépense qu’aurait entraînée la délivrance de la spécialité générique la plus chère du même groupe.
Dans tous les cas, le pharmacien doit inscrire le nom de la spécialité qu’il a délivré sur l’ordonnance.
Le prescripteur peut exclure la substitution générique par le pharmacien dans 3 situations médicales :
- Prescription de médicaments à marge thérapeutique étroite pour assurer la stabilité de la dispensation, lorsque les patients sont effectivement stabilisés avec un médicament, et à l’exclusion des phases d’adaptation du traitement = “non substituable (MTE)”
Principes actifs concernés : lamotrigine, pregabaline, zonisamide, lévétiracétam, topiramate (*), valproate de sodium (*), lévothyroxine, mycophénolate mofétil (*), buprénorphine, azathioprine, ciclosporine, évérolimus, mycophénolate sodique, lacosamide, oxcarbazépine (*) sous TFR.
Sources : arrêté du 12 novembre 2019 , arrêté du 20 juillet 2022
- Prescription chez l’enfant de moins de six ans , lorsqu’aucun médicament générique n’a une forme galénique adaptée et que le médicament de référence disponible permet cette administration = “ non substituable (EFG)”
- Prescription pour un patient présentant une contre-indication formelle et démontrée à un excipient à effet notoire présent dans tous les médicaments génériques disponibles, lorsque le médicament de référence correspondant ne comporte pas cet excipient = “non substituable (CIF)”
Cette mention est indiquée sur l’ordonnance de manière informatisée ou à défaut manuscrite.
Exclusion de la substitution générique par le pharmacien
Les pharmaciens peuvent exclure la substitution générique des médicaments à marge thérapeutique étroite , même lorsque le prescripteur ne l’a pas fait. Le pharmacien doit alors apposer la mention manuscrite « non substituable (MTE-PH) » sur l’ordonnance (le cas échéant pour chaque médicament prescrit) et en informer le prescripteur.
NB : Si la prescription est établie en dénomination commune et qu’il n’existe pas de groupe générique, le pharmacien d’officine dispensera la spécialité adéquate correspondante.
LES BIOSIMILAIRES
Définition
Un médicament biosimilaire, se définit de la façon suivante : « tout médicament biologique de même composition qualitative et quantitative en substance active et de même forme pharmaceutique qu’un médicament biologique de référence mais qui ne remplit pas les conditions prévues au a du 5° du présent article pour être regardé comme une spécialité générique en raison de différences liées notamment à la variabilité de la matière première ou aux procédés de fabrication et nécessitant que soient produites des données précliniques et cliniques supplémentaires … » (article L.5121-1 15° du CSP) . Le médicament biologique est lui-même caractérisé par la présence d’une substance active produite par une source biologique (une bactérie par exemple). Le médicament biosimilaire ne peut être considéré comme un médicament générique d’un médicament biologique, en raison précisément de ce mode de production qui entraîne des variabilités.
Autorisation de mise sur le marché (AMM)
Pour le médicament biosimilaire, en raison des particularités mentionnées précédemment, le dossier de demande d’AMM est plus conséquent et il convient de démontrer la non-infériorité du biosimilaire par rapport au médicament biologique de référence, ainsi que la biosimilarité sur tous les plans du dossier. Quant au prix, il est inférieur d’environ 30 % à celui du médicament de référence.
Répertoire des biosimilaires
Le directeur général de l’ANSM établit un répertoire des médicaments biosimilaires dans lequel figurent les groupes de médicaments biologiques similaires avec les médicaments biologiques de référence et leurs biosimilaires.
Droit de substitution biosimilaire
Les conditions sont les suivantes :
– Le médicament biologique similaire délivré appartient au même groupe biologique similaire que le médicament biologique prescrit ;
– Ce groupe biologique similaire figure sur une liste fixée par arrêté (avis ANSM), accompagnée le cas échéant de conditions de substitution et d’information du prescripteur et du patient.
Aujourd’hui la liste des médicaments substituables ainsi que les conditions de substitution, d’information et des mises en garde de nature à assurer la continuité du traitement sont fixées par cet arrêté (filgrastim,Pegfilgrastim et Ranibizumab).
Le prescripteur informe le patient de la possibilité de substitution par le pharmacien du médicament biologique prescrit ;
Il peut exclure la possibilité de cette substitution par une mention expresse et justifiée portée sur l’ordonnance, tenant à la situation médicale du patient.
En cas de substitution, le pharmacien :
- informe le patient lors de la dispensation de la substitution effective et des informations utiles associées ;
- mentionne sur l’ordonnance le nom du médicament effectivement dispensé ;
- informe le prescripteur quant au médicament dispensé ;
- procède à l’enregistrement du nom du médicament délivré par substitution et son numéro de lot par tous moyens adaptés afin de mettre en œuvre la traçabilité requise pour tous les médicaments biologiques ;
- assure la continuité de la dispensation du même médicament lors des dispensations suivantes.
– Cette substitution ne doit pas entraîner une dépense supplémentaire pour l’assurance maladie supérieure à la dépense qu’aurait entraînée la délivrance du médicament biologique similaire le plus onéreux du même groupe.
Lorsqu’un grand conditionnement est disponible pour la forme biologique similaire du médicament et que le traitement en est prescrit pour une durée d’au moins trois mois, y compris par renouvellement multiple d’un traitement mensuel, le pharmacien délivre un grand conditionnement.
Source: L 5125-23-2 du CSP
MÉDICAMENTS HYBRIDES
Définition
Une spécialité hybride d’une spécialité de référence, est une spécialité contenant le même principe actif mais qui ne répond pas à la définition d’une spécialité générique parce qu’elle comporte par rapport à la spécialité de référence des différences relatives:
- aux indications thérapeutiques
- au dosage,
- à la forme pharmaceutique
- à la voie d’administration
- ou lorsque la bio-équivalence par rapport à cette spécialité de référence n’a pu être démontrée par des études de biodisponibilité.
L’autorisation de mise sur le marché (AMM)
L’autorisation de mise sur le marché d’une spécialité hybride repose au moins pour partie sur les résultats des essais pré-cliniques et cliniques appropriés déterminés en fonction de ces différences.
Le registre des hybrides
Le registre des groupes hybrides comporte les groupes dans lesquels figurent une spécialité de référence et des spécialités qui en sont hybrides.
Aujourd’hui, il comporte uniquement les médicaments hybrides relevant des classes des médicaments des maladies obstructives des voies respiratoires (administrés par voie inhalée): INNOVAIR, FLIXOTIDE 125 ET 250, SERETIDE 125 etc.
Consultez le registre des hybrides.
La substitution hybride
Le pharmacien peut délivrer, par substitution à la spécialité prescrite, une spécialité du même groupe hybride, à condition que le prescripteur n’ait pas exclu cette possibilité par une mention expresse et justifiée portée sur l’ordonnance.
L’ arrêté du 31 janvier 2023 précise les situations médicales dans lesquelles cette exclusion peut être justifiée. Il s’agit :
- d’une prescription chez l’enfant de moins de six ans, lorsqu’aucune spécialité du même groupe hybride inscrits au registre des groupes hybrides n’a une forme galénique adaptée et que la spécialité de référence disponible permet cette administration (mention à reporter sur l’ordonnance « non substituable (EFG) »)
- d’une prescription pour un patient présentant une contre-indication formelle et démontrée à un excipient à effet notoire présent dans toutes les spécialités disponibles du même groupe hybride inscrits au registre des groupes hybrides lorsque la spécialité de référence correspondante ne comporte pas cet excipient (mention à reporter sur l’ordonnance « non substituable (CIF) »).
Lors de la délivrance d’un médicament hybride d’un médicament de référence, le pharmacien informe le patient de la substitution du médicament de référence prescrite par un médicament du même groupe hybride.
Source : article L5125-23 du CSP
Considérations déontologiques
L’accès du public aux médicaments
C’est parce que le médicament est un bien particulier que le Code de déontologie impose au pharmacien de veiller à ce que le public ne puisse pas y accéder directement (R. 4235-55 du CSP).
Seuls certains médicaments (clairement définis sur le site de l’ANSM et de MEDDISPAR) dits de « médication officinale » peuvent être mis à disposition du public en « libre accès » dans les pharmacies d’officine (c’est à dire devant les comptoirs).
Ces médicaments doivent être présentés dans un espace dédié, clairement identifié et situé à proximité immédiate des postes de dispensation des médicaments et d’alimentation du dossier pharmaceutique, de façon à permettre un contrôle effectif du pharmacien. Ils ne sont pas mêlés à d’autres produits ayant un autre statut juridique (ex: compléments alimentaires, produits cosmétiques …).
Le pharmacien doit, en plus de son rôle d’accompagnement et de conseil auprès du patient, mettre à la disposition du public les informations émanant des autorités de santé relatives au bon usage de ces médicaments.
La liste évolue régulièrement. Pour exemple, en 2020, les médicaments à base de paracétamol ou d’anti-inflammatoires non stéroïdiens ont été retirés du libre accès.
L’incitation à la consommation abusive de médicaments est interdite
Le pharmacien ne doit pas, par quelque procédé ou moyen que ce soit, inciter ses patients à une consommation abusive de médicaments (R. 4235-64 du CSP). Par exemple, une incitation promotionnelle du type “3 pour le prix de 2” d’un médicament non remboursable constitue un manquement à la déontologie.
En effet, proposer un prix dégressif en fonction de la quantité achetée constitue une promotion commerciale que la spécificité du médicament interdit.
Lutte contre le charlatanisme
Le pharmacien doit contribuer à la lutte contre le charlatanisme, notamment en s’abstenant de fabriquer, distribuer ou vendre tous objets ou produits ayant ce caractère (R. 4235-10 du CSP).
La publicité concernant les médicaments
La publicité pour les médicaments à destination des professionnels de santé et du grand public est soumise à un visa délivré par l’ANSM.
Le contrôle effectué à priori par l’ANSM comporte les critères suivants :
- Respecter les dispositions de l’autorisation de mise sur le marché et les stratégies thérapeutiques recommandées par la Haute Autorité de Santé ;
- Présenter le médicament de façon objective et favoriser son bon usage ;
- Ne pas être trompeuse, ni porter atteinte à la protection de la santé publique.
Lorsqu’un médicament fait l’objet d’une réévaluation du rapport bénéfice/risque, sa publicité est interdite jusqu’à l’issue de cette procédure.
Publicité auprès des professionnels de santé – visa PM
La publicité doit être conforme à l’autorisation de mise sur le marché et reproduire le résumé des caractéristiques du produit (RCP) qui mentionne notamment les effets indésirables, les contre-indications ainsi que les interactions du médicament considéré.
Toute publicité auprès des professionnels de santé pour des vaccins est assortie, de façon clairement identifiée et sans renvoi, des recommandations in extenso de l’avis de la Haute Autorité de Santé.
Publicité auprès du grand public – visa GP
Elle est autorisée uniquement pour les médicaments :
- Non soumis à prescription médicale obligatoire ;
- Non remboursables par les régimes obligatoires d’assurance maladie (pour aucune de ses différentes présentations).
De plus, leur AMM ne doit pas faire état de restrictions en matière de publicité « grand public ». Deux exceptions demeurent : les vaccins et les médicaments de sevrage tabagique pour des raisons de santé publique.
La vente en ligne de médicaments est soumise aux mêmes règles.
Le pharmacien d’officine peut utiliser uniquement les supports publicitaires portant un visa GP dans son officine ou sa vitrine (PLV, affiche, film, etc.).
Falsification et contrefaçon de médicaments
La production et la vente de médicaments falsifiés constituent un commerce illégal d’envergure mondiale en pleine croissance.
Au niveau mondial, les médicaments falsifiés ont été reconnus comme une menace pour la santé publique par l’OMS qui estime par ailleurs qu’environ 50 % des médicaments vendus sur Internet sont des médicaments falsifiés (médicaments contrefaits, médicaments non autorisés…).
Dispositions européennes
Le Conseil de l’Europe a mis en place en 2010 le 1er traité international sur les produits de santé contrefaits ou falsifiés menaçant la Santé Publique : la « Convention Medicrime ». Ce traité offre un cadre juridique aux Etats Membres pour qu’ils prennent toutes les mesures dissuasives nécessaires.
En juin 2011, l’Union européenne a adopté la directive 2011/62/UE relative à la prévention de l’introduction dans la chaîne d’approvisionnement légale de médicaments falsifiés conduisant notamment à la sérialisation de certains médicaments.
Opérations internationales “PANGEA”
Depuis plusieurs années, des actions internationales de grande ampleur, telles que les opérations PANGEA, sont menées régulièrement pour lutter notamment contre la vente illicite de médicaments par internet. Ces opérations internationales sont coordonnées par Interpol et l’Organisation mondiale des douanes.
La dernière opération en date, PANGEA XIII est destinée à lutter contre la vente en ligne de médicaments et de dispositifs médicaux illicites et contrefaits. L’opération a été conduite du 3 au 10 mars 2020 simultanément dans 90 pays.
Au niveau international, 4,4 millions d’unités de produits pharmaceutiques illicites ont été saisies dans le monde, dont des comprimés contre les troubles de l’érection, des médicaments anticancéreux, hypnotiques et sédatifs, des stéroïdes anabolisants, des analgésiques… Plus de 37 000 dispositifs médicaux non autorisés et contrefaits ont également été saisis, principalement des masques chirurgicaux et des autotests pour dépister le VIH ou mesurer la glycémie. L’opération a permis de procéder à 121 arrestations dans le monde entier. Les malfaiteurs ont particulièrement profité de l’importante demande de produits de protection individuelle et d’hygiène personnelle dans le cadre de la crise sanitaire liée au Covid-19.
Par ailleurs, plus de 2 500 liens renvoyant vers des sites web, des médias sociaux, des places de marché électronique ou encore des publicités en ligne pour des produits pharmaceutiques illicites, ont été supprimés.
Les dispositions françaises
Conditions de mise sur le marché des médicaments en France : un triple régime d’autorisation (source ANSM)
Chaque spécialité pharmaceutique fait l’objet d’une Autorisation de Mise sur le Marché (AMM) garantissant sa qualité, sa sécurité et son efficacité. L’ensemble de la chaîne pharmaceutique d’importation, de fabrication, de distribution en gros et de vente au détail par les pharmacies d’officine et les pharmacies à usage intérieur d’établissements de santé est soumise à des obligations :
- Les établissements pharmaceutiques sont autorisés par l’ANSM ;
- Les établissements de distribution au détail sont autorisés par les ARS (cas des pharmacies d’officine).
Ces établissements sont placés sous la responsabilité de pharmaciens inscrits à l’Ordre national des pharmaciens. Ce dispositif, complété par des inspections régulières, participe à la lutte contre les médicaments falsifiés. Tout médicament importé en Europe doit faire l’objet d’un contrôle de la qualité du produit fini à son arrivée sur le territoire national. Ce contrôle est effectué par l’établissement pharmaceutique importateur. Le respect de cette obligation réglementaire contribue à prévenir l’introduction de médicaments falsifiés au sein de l’Europe.
Les sanctions prévues en France
La fabrication, le courtage, la distribution, la publicité, l’offre de vente, la vente, l’importation et l’exportation de médicaments falsifiés (ou tentative de) sont punis de cinq ans d’emprisonnement et de 375 000 euros d’amende (portés à sept ans d’emprisonnement et 750 000 euros d’amende lorsque le médicament falsifié est dangereux pour la santé de l’homme ; les délits ont été commis par des professionnels autorisés ; les délits ont été commis en bande organisée ; les délits de publicité, offre de vente ou vente de médicaments falsifiés ont été commis sur un réseau de télécommunication à destination d’un public non déterminé).
Ceux qui, sans motif légitime, sont trouvés en France détenteurs de médicaments falsifiés, sont punis de trois ans d’emprisonnement et de 75 000 euros d’amende. Lorsque le médicament falsifié est dangereux pour la santé de l’homme, ces peines sont portées à cinq ans d’emprisonnement et à 375 000 euros d’amende.
En savoir plus: La lutte contre les médicaments falsifiés (ONP)